Abordagem diagnóstica da anemia hemolítica
Pontos principais do artigo
A anemia hemolítica não é uma doença única, mas, na verdade, um grupo de condições em que se identifica uma destruição aumentada (precoce) de hemácias que supera a capacidade compensatória da medula óssea.
Como a vida média normal da hemácia é de aproximadamente 120 dias, a hemólise representa uma redução anormal desta duração e geralmente é acompanhada de reticulocitose.
Do ponto de vista fisiopatológico, a hemácia pode ser destruída na circulação (hemólise intravascular) ou no sistema reticuloendotelial, sobretudo no baço e no fígado (hemólise extravascular).
Na hemólise extravascular, o mecanismo central é a retenção e fagocitose de hemácias pouco deformáveis, incapazes de atravessar adequadamente os cordões esplênicos.
Isso ocorre por alterações da membrana, da hemoglobina ou do metabolismo eritrocitário, como nas membranopatias, hemoglobinopatias e enzimopatias.
Já a hemólise intravascular costuma decorrer de lesão mais direta da hemácia, por complemento, toxinas, trauma mecânico, fragmentação em microvasos ou estresse oxidativo.
Nas anemias microangiopáticas, por exemplo, a passagem por vasos com microtrombos leva à fragmentação das hemácias; de outro lado, em pacientes com válvulas protéticas, este fenômeno é primariamente mecânico.
As anemias hemolíticas também podem ser classificadas como hereditárias ou adquiridas, agudas ou crônicas, imunes ou não imunes, e por defeitos intrínsecos ou extrínsecos à hemácia.
Em geral, defeitos intrínsecos são hereditários e defeitos extrínsecos são adquiridos, mas há exceções importantes: a hemoglobinúria paroxística noturna é adquirida, embora decorra de uma alteração intrínseca da hemácia; já a deficiência de G6PD é hereditária, mas frequentemente se manifesta após um gatilho externo.
Identificando hemólise
O primeiro passo no processo diagnóstico de anemia hemolítica é identificar o achado “síndrômico": a presença de hemólise. Para isso, a avaliação laboratorial é central.
A hemólise se caracteriza pelo aumento da degradação da hemoglobina e pela resposta compensatória da medula óssea.
Assim,
os principais achados são anemia associada a reticulocitose, aumento de LDH (lactato desidrogenase), elevação de bilirrubina indireta e redução da haptoglobina.
A reticulocitose reflete a tentativa da medula de compensar a perda periférica de hemácias; por isso, sua ausência não exclui hemólise quando há deficiência de ferro, folato ou vitamina B12, doença renal crônica, inflamação ou comprometimento medular.
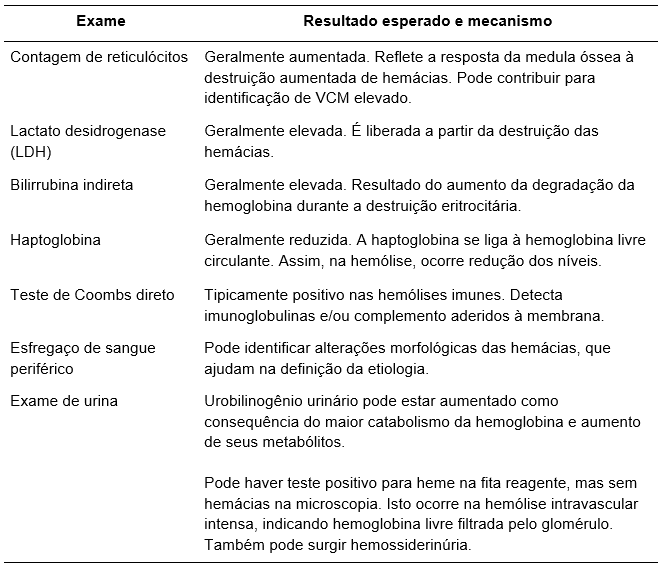
A
Tabela 1
lista os exames para o diagnóstico de hemólise e seu mecanismo de alteração.
Tabela 1.
Principais exames na anemia hemolítica. Adaptada das referências 2 e 4.
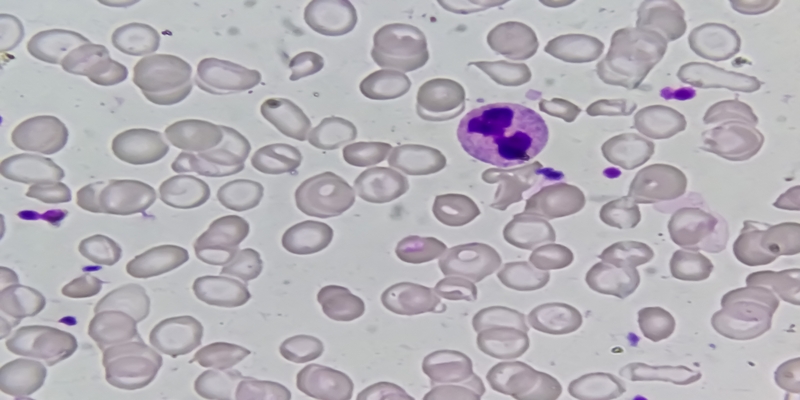
Alguns achados ajudam a sugerir o local predominante da hemólise. Na hemólise intravascular, especialmente quando intensa, a haptoglobina pode se esgotar, permitindo o aparecimento de hemoglobina livre no plasma e na urina, com hemoglobinúria e urina escurecida.
Em episódios mais prolongados, pode haver hemossiderinúria. Já na hemólise extravascular, a destruição das hemácias ocorre sobretudo no baço e no fígado, com maior tendência à icterícia, à esplenomegalia e ao aumento de urobilinogênio.
Na prática, porém, os mecanismos podem se sobrepor, e a distinção nem sempre é absoluta. Apesar da importância do laboratório, o exame clínico não deve ser esquecido.
Devem ser valorizados sintomas
gerais de anemia (fadiga, dispneia, palpitações e palidez) e também achados que apontem para destruição aumentada de hemácias, como icterícia e colúria.
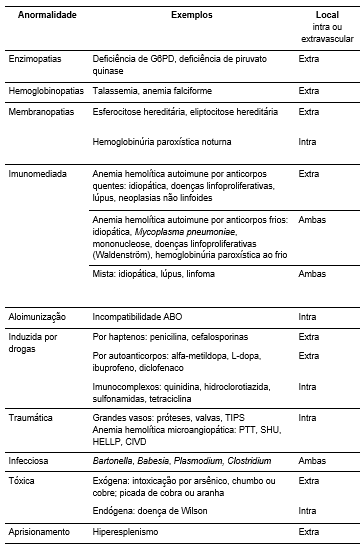
Abordagem diagnóstica
Uma vez confirmada a hemólise, o próximo passo é identificar sua causa (
Tabela 2
). A abordagem deve considerar a gravidade clínica, a história, o exame físico, o hemograma, o esfregaço de sangue periférico e o teste de Coombs (antiglobulina direta).
Tabela 2.
Causas selecionadas de anemia hemolítica. Traduzido e adaptado da referência 3.
O primeiro cuidado é reconhecer situações potencialmente graves e ameaçadoras à vida. Anemia de instalação rápida, hemoglobina muito baixa, instabilidade hemodinâmica, hemoglobinúria intensa, plaquetopenia, esquizócitos no esfregaço ou disfunção renal devem chamar atenção para diagnósticos urgentes, especialmente microangiopatias trombóticas, coagulação intravascular disseminada e reação transfusional aguda.
A presença de esquizócitos e fragmentos eritrocitários no exame periférico sempre deve gerar atenção para a potencial gravidade. Nesses casos, a investigação etiológica não deve atrasar medidas de suporte e avaliação especializada.
A história e o exame físico ajudam a direcionar a investigação. Diarreia recente pode apontar para síndrome hemolítico-urêmica; transfusão recente sugere reação transfusional; sintomas desencadeados pelo frio levantam a possibilidade de doença por aglutininas frias; febre pode acompanhar infecções, anemia hemolítica autoimune, CIVD ou microangiopatias.
Também devem ser pesquisados medicamentos, toxinas, infecções recentes, doenças autoimunes, neoplasias, linfadenomegalias e hepatomegalia.
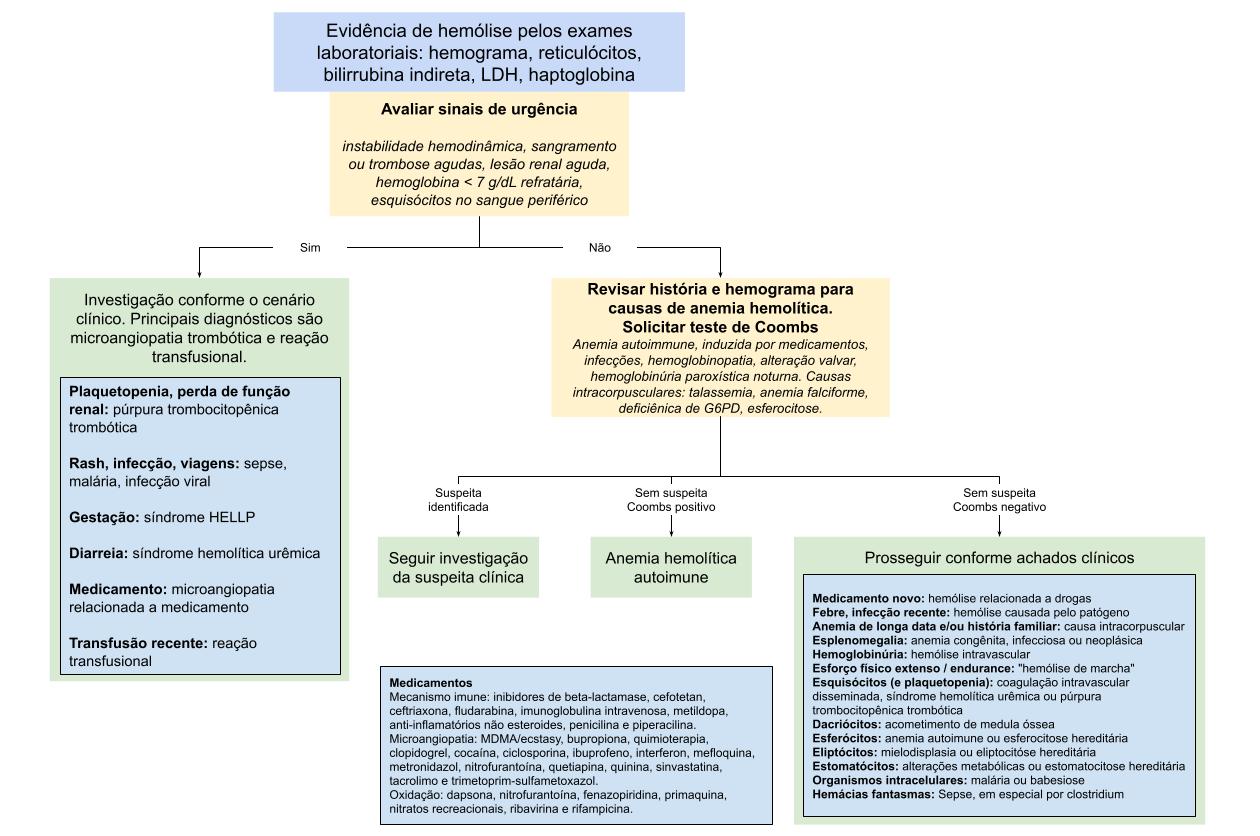
Após essa avaliação inicial, uma divisão prática é separar as hemólises em imunes e não imunes. Este é o principal passo, conforme apresentado na
Figura 1
.
O teste de antiglobulina direta, ou Coombs direto, é central nessa etapa, pois identifica imunoglobulinas ou complemento aderidos às hemácias.
Um teste positivo sugere hemólise imune, como anemia hemolítica autoimune, reação transfusional ou hemólise induzida por medicamentos. O resultado, porém, deve sempre ser interpretado no contexto clínico.
Figura 1.
Abordagem diagnóstica da anemia hemolítica. Adaptada das referências 1 e 2.
Quando o Coombs é negativo, a investigação se apoia especialmente no esfregaço de sangue periférico, na história familiar e no contexto clínico.
Esquizócitos sugerem fragmentação eritrocitária, como nas microangiopatias, CIVD ou próteses valvares. Esferócitos podem ocorrer na esferocitose hereditária e também em hemólise imune.
Hemácias falciformes sugerem doença falciforme; eliptócitos apontam para eliptocitose hereditária; bite cells e corpúsculos de Heinz sugerem hemólise oxidativa, como na deficiência de G6PD.
Se houver suspeita de doença hereditária, a investigação pode seguir com exames específicos, como eletroforese de hemoglobina, testes de membrana eritrocitária ou dosagem de atividade enzimática.
Se o quadro parecer adquirido, devem ser consideradas causas mecânicas, infecciosas, tóxicas, medicamentosas, microangiopáticas e sistêmicas.
Assim, a sequência prática é: confirmar hemólise, reconhecer gravidade, buscar pistas clínicas, realizar Coombs direto e interpretar o esfregaço periférico.
Situações especiais
A relação entre hemólise, anemia e reticulocitose nem sempre é linear. Em alguns pacientes, pode haver hemólise sem anemia, quando a medula óssea consegue aumentar a produção de hemácias o suficiente para compensar a destruição periférica.
Este cenário se apresenta com hemoglobina normal, mas com os marcadores de hemólise alterados.
Nestes casos, a conduta é reconhecer que há hemólise ativa e prosseguir a investigação etiológica, mesmo sem anemia.
O contrário também exige atenção: reticulocitose não indica de forma automática a presença de hemólise.
Ela pode ocorrer após sangramento recente, durante a recuperação de uma deficiência de micronutrientes (ferro, vitamina B12, folato), após uso de eritropoetina ou na recuperação de uma agressão medular transitória.
Em suma, deve-se revisar o contexto clínico e confirmar se os demais marcadores de hemólise estão presentes antes de rotular o quadro como anemia hemolítica.
Por fim, a hemólise pode ocorrer sem reticulocitose. Isso costuma indicar resposta medular inadequada à hemólise, seja por condição concomitante, seja por atraso transitório nos primeiros dias do quadro.
As causas desse fenômeno são variadas e incluem deficiência nutricional, doença renal crônica, inflamação, álcool, mielodisplasia e supressão medular por medicamentos.
Raramente a anemia hemolítica autoimune pode ter acometimento de precursores eritroides. Portanto, deve-se integrar hemoglobina, reticulócitos, marcadores de hemólise e contexto clínico.