Fenilcetonúria (PKU): diagnóstico precoce e abordagens terapêuticas atualizadas
A fenilcetonúria (FNC) é uma doença autossômica recessiva, causada por mutações no gene PAH que codifica a enzima hepática fenilalanina-hidroxilase (FAH). A ausência (ou atividade deficiente) dessa enzima impede a conversão de fenilalanina em tirosina, causando o acúmulo de fenilalanina no sangue e no líquor.
É uma doença metabólica rara, com prevalência global média estimada de 1:10.000 recém-nascidos. A incidência varia entre as diferentes nações do mundo e os diferentes grupos étnicos. As maiores taxas são encontradas na Irlanda (1: 4.500) e na Turquia (1: 2.600), e as menores, na Finlândia, no Japão e na Tailândia (1:200.000, 1: 143.000 e 1: 212.000, respectivamente). No Brasil, tem sido encontrada uma prevalência variando de 1: 15.000 a 1: 25.000.
Esse quadro pode gerar um efeito neurotóxico, levando a problemas no desenvolvimento neuromotor e neurocognitivo. A triagem neonatal no Brasil é feita através do teste do pezinho na primeira semana de vida. A fenilcetonúria (PKU), quando não tratada, pode levar a diversos problemas de saúde, principalmente relacionados ao desenvolvimento neurológico. É importante notar que o tratamento precoce, especialmente através da restrição dietética de Phe, pode prevenir estas sequelas e permitir que indivíduos com PKU tenham desenvolvimento intelectual normal.
Definição e etiologia da PKU
A fenilcetonúria (PKU), também conhecida como deficiência de fenilalanina hidroxilase (PAH), é uma doença autossômica recessiva do metabolismo da fenilalanina (Phe). Nesta condição, concentrações elevadas de Phe, especialmente no cérebro, causam disfunção cerebral.
A PKU é causada por variantes patogênicas no gene PAH, responsável pela conversão de Phe em tirosina (Tyr). A reação de conversão de Phe para Tyr requer a co-substrato tetrahidrobiopterina (BH4). Defeitos no metabolismo do BH4 também podem causar hiperfenilalaninemia (HPA) em alguns casos. A deficiência da atividade da enzima PAH leva à acumulação de Phe no sangue (HPA), com níveis normais de Phe no sangue a rondar os 35–120 μmol/l.
Em doentes não tratados com PKU, as concentrações de Phe no sangue são marcadamente aumentadas, resultando na formação de corpos de fenilcetona que são excretados na urina. As concentrações de Tyr são normalmente baixas.
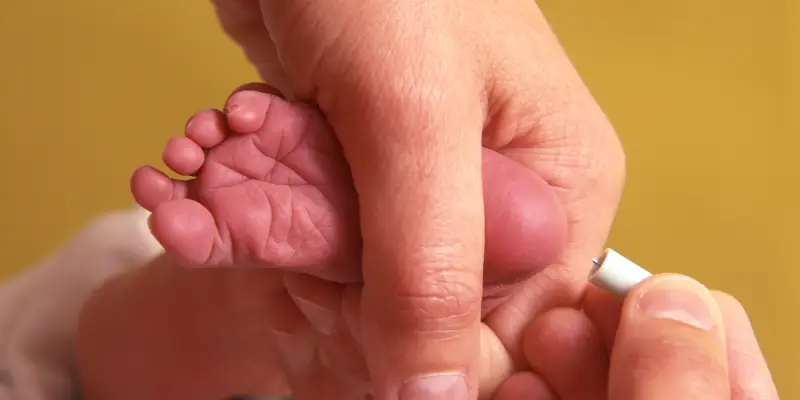
Diagnóstico precoce: papel do teste do pezinho
A triagem neonatal no Brasil é feita através do teste do pezinho na primeira semana de vida. O teste do pezinho, como o próprio nome diz, é um teste de triagem, não sendo, portanto, definidor de diagnóstico e sim um indicador de que são necessárias maiores investigações.
Quando a dosagem de fenilalanina no exame do “Teste do Pezinho” (Triagem Neonatal) for menor que 117 mcmol/L, o resultado é considerado normal.
Quando a dosagem de fenilalanina for maior ou igual a 117 mcmol/L, mas menor que 480 mcmol/L, será solicitada nova coleta de sangue para se fazer outra dosagem de fenilalanina. Nesse caso, há duas possibilidades:
- Se o resultado for menor que 117 mcmol/L, o exame é considerado normal e a criança terá, portanto, uma triagem negativa para fenilcetonúria.
- Se o resultado for igual ou maior que 117 mcmol/L, a criança deverá realizar novos exames para confirmar ou descartar a doença.
Quando a dosagem de fenilalanina, no exame de triagem neonatal, for maior ou igual a 480 mcmol/L, a criança tem maior possibilidade de ter fenilcetonúria, devendo ser submetida a novos exames e consulta médica precoce.
Diagnóstico diferencial com outras doenças metabólicas
O diagnóstico diferencial da fenilcetonúria (PKU) com outras doenças metabólicas é essencial quando se detecta hiperfenilalaninemia (HPA) no rastreio neonatal. A HPA, que é o principal achado bioquímico da PKU, pode ser causada por diferentes defeitos metabólicos, sendo essencial distinguir entre eles para um tratamento adequado.
- Deficiência de PAH (Fenilalanina Hidroxilase):a PKU é causada por variantes patogênicas no genePAH, que codifica a enzima fenilalanina hidroxilase. Esta enzima é responsável pela conversão de fenilalanina (Phe) em tirosina (Tyr). A deficiência na atividade desta enzima leva à acumulação de Phe no sangue.
- Defeitos no metabolismo da tetrahidrobiopterina (BH4):o BH4 é um cofator essencial para a atividade da PAH. Defeitos no metabolismo do BH4 podem causar HPA. Existem diferentes causas genéticas para estes defeitos, incluindo deficiências em: GTPCH (GTP ciclohidrolase), PTPS (6-piruvoil tetrahidropterina sintase), DHPR (dihidropteridina redutase) ou SR (sepiapterina redutase).
- Deficiência de DNAJC12:a proteína chaperona DNAJC12 facilita o correto dobramento do monômero de PAH. Variantes patogênicas neste gene também podem causar HPA.
Sinais e sintomas
A criança com fenilcetonúria, sem tratamento, somente começa a apresentar sintomas e sinais evidentes da doença entre os três e seis meses de vida.
As manifestações mais frequentes são:
- Vômitos frequentes;
- Problemas de pele (descamação, vermelhidão);
- Cheiro diferente, mais forte na urina;
- Microcefalia (cabeça com tamanho menor que o normal);
- Convulsões (acessos);
- Músculos rígidos, “duros”, difíceis de “esticar”;
- Hiperatividade (muita agitação);
- Atraso do crescimento, não ganha peso e não cresce direito;
- Atraso do desenvolvimento (atraso para sorrir, firmar a cabeça, sentar) ou regressão do desenvolvimento – deixa de fazer algumas das etapas que já havia adquirido – como sorrir, firmar a cabeça, olhar o rosto das pessoas, sentar, entre outras.
Os impactos da PKU não tratada incluem:
- Deficiência intelectual grave:a acumulação de fenilalanina (Phe) no cérebro leva à disfunção cerebral, resultando em grave deficiência intelectual se não tratada
- Problemas neurológicos:pacientes não tratados podem desenvolver epilepsia, problemas comportamentais, psiquiátricos e de movimento
- Problemas de pigmentação:a falta de tratamento pode levar a uma pigmentação mais clara da pele, olhos e cabelo
- Eczema e odor:pode ocorrer eczema e um odor característico de mofo no corpo de indivíduos não tratados
Além desses impactos principais, a PKU não tratada pode acarretar outros problemas de saúde, embora com menor frequência:
- Cegueira cortical:em alguns casos, a falta de tratamento pode levar à cegueira cortical.
- Problemas de humor, memória e função executiva:adultos com PKU que não seguem o tratamento podem ter problemas de atenção, humor, memória e função executiva.
- Espasticidade e convulsões:em alguns casos, pacientes que não aderem ao tratamento a longo prazo podem desenvolver espasticidade dos membros inferiores e convulsões.
- Encefalopatia:em casos de falta de controle metabólico prolongada, pode ocorrer encefalopatia, que é reversível com tratamento dietético rigoroso.
- Anormalidades visuais:problemas visuais podem surgir em adultos que não seguem o tratamento.
Abordagens terapêuticas atualizadas
O tratamento primário para PKU é uma dieta restrita em Phe, que inclui o uso de uma fórmula especial (sem Phe), suplementação de tirosina e outros nutrientes essenciais e alimentos com baixo teor de proteína.
Além da dieta, existem tratamentos farmacológicos como o uso de sapropterina dicloridrato (análogo sintético da BH4), que pode aumentar a tolerância à Phe em pacientes BH4-responsivos. Em casos mais severos, uma terapia enzimática de substituição com pegvaliase também pode ser utilizada para reduzir as concentrações de Phe.
Novas abordagens como terapia genética, terapias com mRNA e terapia com células vermelhas do sangue carregadas com a enzima fenilalanina amônia liase (PAL) estão em desenvolvimento.
A sapropterina dicloridrato (BH4) é uma forma sintética de tetrahidrobiopterina, um cofator da PAH. Ela funciona estabilizando a estrutura da PAH, aumentando a atividade enzimática residual em pacientes com certas mutações.
O tratamento com BH4 é mais eficaz em pacientes com PKU BH4-responsiva, que mostram uma diminuição nos níveis de Phe quando tomam o medicamento. A BH4 pode permitir que esses pacientes tenham uma dieta menos restritiva, em comparação com a dieta apenas, mas é menos eficaz em pacientes BH4-não responsivos.
Desafios no acompanhamento
A adesão à dieta restrita em Phe pode ser um desafio, especialmente em adolescentes e adultos, devido às restrições alimentares e à necessidade de um controle rigoroso. Isso pode afetar a qualidade de vida, incluindo a saúde mental, bem-estar psicológico e vida social.
Apesar desses desafios, a qualidade de vida de muitos pacientes com PKU bem controlados pode ser comparável à da população geral. Abordagens como suporte psicológico, programas educacionais, aplicativos e tratamentos alternativos podem ajudar a melhorar a adesão e o bem-estar.
Conclusão
A fenilcetonúria (PKU) é um distúrbio metabólico hereditário causado pela deficiência da enzima fenilalanina hidroxilase (PAH), resultando em níveis elevados de fenilalanina (Phe) no sangue e no cérebro, o que pode levar a disfunções neurológicas graves se não tratada.
O diagnóstico precoce é fundamental e é geralmente realizado através de programas de triagem neonatal, que têm sido eficazes na identificação de casos de PKU logo após o nascimento. O tratamento da PKU tradicionalmente envolve uma dieta rigorosa com restrição de fenilalanina, suplementada por alimentos médicos especiais que contêm outros aminoácidos essenciais.
No entanto, mesmo com o tratamento, alguns pacientes podem apresentar alguns problemas neurocognitivos, particularmente se o controle metabólico não for mantido de forma consistente, especialmente durante a adolescência e idade adulta. Além disso, a má adesão ao tratamento pode levar a problemas psicológicos e dificuldades sociais, impactando a qualidade de vida.