Fibrose cística para além do teste do pezinho: como realizar o diagnóstico?

Pontos principais do artigo
A fibrose cística (FC) é uma doença genética autossômica recessiva caracterizada pela disfunção do gene CFTR. Trata-se de uma doença multissistêmica que ocorre mais frequentemente em populações descendentes de caucasianos. Nas últimas décadas, diversos avanços no diagnóstico e tratamento da FC mudaram drasticamente o cenário dessa doença, com aumento expressivo da sobrevida e qualidade de vida.
Atualmente, o Brasil dispõe de um programa de ampla cobertura para a triagem neonatal de FC e centros de referência distribuídos na maior parte desses estados para seguimento dos indivíduos. Antigamente, confinada à faixa etária pediátrica, tem-se observado um aumento de pacientes adultos com FC tanto pelo maior número de diagnósticos de formas atípicas, de expressão fenotípica mais leve, assim como pelo aumento da expectativa de vida com os novos tratamentos. Entretanto, ainda se observa uma grande heterogeneidade no acesso aos métodos diagnósticos e terapêuticos para FC entre as diferentes regiões brasileiras.
No Brasil, estima-se que a incidência de fibrose cística seja de 1:7.576 nascidos vivos; porém, apresenta diferenças regionais, com valores mais elevados nos estados da região Sul. Atualmente, existem quase 2.700 brasileiros com fibrose cística.
Diagnóstico clínico
Entre as consequências da FC está a produção de muco exageradamente espesso, que não é devidamente eliminado pelo organismo. O acúmulo de muco nas vias do corpo e em órgãos – como pulmões, pâncreas, fígado e intestino – provoca alterações no funcionamento destes órgãos.
Nos pulmões, este acúmulo de secreção associa-se ao aparecimento de bactérias que causam infecções graves e constantes, causando inflamação pulmonar e colocando a saúde dos pacientes em risco. Além da
Pseudomonas aeruginosa
, outras bactérias comuns nas vias aéreas são
Staphylococcus aureus
e
Haemophilus infuenzae
.
A manifestação e a gravidade dos sintomas da FC variam entre cada paciente e as fases da vida. Os sistemas respiratório, digestivo e reprodutivo são normalmente os mais afetados pelo acúmulo de muco espesso na FC, por isso, os mais impactados pelos sintomas da doença.
Os sintomas comuns da FC em crianças são:
- dificuldade de crescimento, ganho de peso abaixo da normalidade, deficiência de vitaminas e desnutrição;
- fezes anormalmente volumosas, gordurosas e com mau cheiro;
- inchaço na barriga, constipação grave, diarreia, dor e desconforto abdominais
- pneumonias e bronquites frequentes;
- sinusite crônica;
- tosses constantes com catarro e eventualmente sangue;
- pele com gosto salgado, decorrente do excesso de sal no suor;
- aparecimento de pólipos nasais (tecido inflamado dentro do nariz);
- infecções respiratórias frequentes causadas pelas bactériasPseudomonas aeruginosa(Pa),Staphylococcus aureusouHaemophilus infuenzae.
Na vida adulta, o avanço da doença, além das alterações pulmonares decorrentes de infecções recorrentes, pode causar dificuldade reprodutiva por azoospermia em homens e obstrução do colo do útero em mulheres, além de osteopenia e osteoporose, baqueteamento digital e diabetes. É importante ressaltar que, por se tratar de uma doença genética, o filho de uma pessoa com fibrose cística teria maior probabilidade de também nascer com a doença: a chance é de no mínimo 50%.
Testes diagnósticos
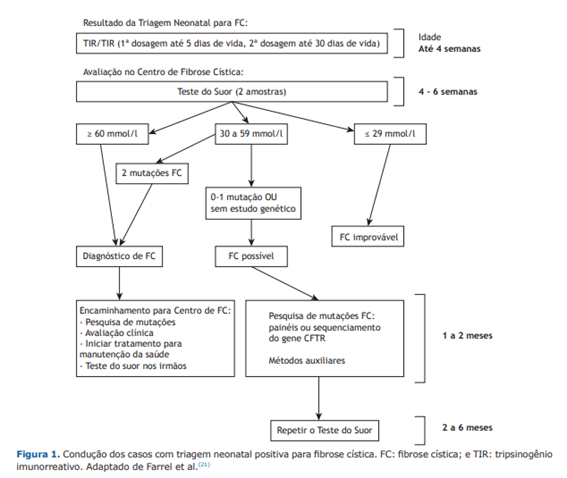
O diagnóstico da fibrose cística é estabelecido quando um paciente com sintomas sugestivos ou um teste de triagem neonatal alterado tem duas dosagens de cloro no suor alteradas (acima de 60 mEq/L) ou duas mutações identificadas no teste genético.
Triagem neonatal (teste do pezinho)
A triagem neonatal consiste na retirada de algumas gotas de sangue do calcanhar do bebê, na primeira semana de vida. O teste de triagem neonatal não é um exame definitivo, ou seja, ele não determina se o bebê tem ou não fibrose cística, mas é capaz de indicar a necessidade de exames complementares para definir se o paciente tem de fato a doença. A realização do teste do pezinho é fundamental, pois permite realizar o diagnóstico antes do aparecimento dos primeiros sintomas da doença, melhorando muito o prognóstico dos pacientes.
Teste do suor
É realizado apenas quando há suspeita da fibrose cística, através da indução e coleta do suor. O teste avalia a concentração de cloro (ou cloreto) no suor, que acima de um determinado nível indica a existência da doença (60 mEq/L ou mais, dependendo da técnica utilizada).
Teste genético
É uma opção para o diagnóstico de pessoas com suspeita de fibrose cística que apresentam níveis intermediários de cloreto no suor (entre 40 e 60 mEq/L) ou mesmo níveis normais. Além disso, é útil para determinar o prognóstico dos pacientes, auxiliar no aconselhamento familiar e determinar possibilidades de tratamento com novos medicamentos que “corrigem” a função da proteína CFTR.
Nos casos em que a mãe do bebê tem fibrose cística ou quando há casos na família, recomenda-se também a investigação diagnóstica, através de testes de suor ou testes genéticos. Utilizando o teste genético também é possível realizar diagnóstico pré-natal em alguns casos, ou seja, antes do nascimento do bebê.
O diagnóstico pré-natal para avaliar se o feto possui fibrose cística é realizado apenas se exames prévios confirmarem que tanto o pai quanto a mãe do bebê carregam o gene afetado pela doença (CFTR). Isso porque o bebê apenas poderá apresentar a FC se herdar o gene afetado tanto do pai quanto da mãe. O exame pré-natal consiste na retirada de uma amostra do líquido amniótico do útero para determinar o diagnóstico da fibrose cística. Este exame é geralmente realizado entre a 15ª e a 18ª semana da gestação.
Mesmo quando os pais não sabem que carregam o gene afetado pela FC, a triagem da doença deve idealmente ocorrer logo nos primeiros dias de vida, através do teste do pezinho (triagem neonatal).
Abaixo, algoritmo sugerido para diagnóstico de FC:
Desafios no diagnóstico precoce
Apesar da triagem neonatal ser um exame obrigatório no Brasil, seu acesso ainda não é universal no país. Por isso, ainda existem casos em que o diagnóstico é feito de forma tardia, após o aparecimento dos sintomas da doença. Esse atraso no diagnóstico da fibrose cística pode comprometer o quadro geral de saúde do paciente e seu prognóstico.
Em áreas com recursos limitados, o teste de cloreto no suor pode não estar disponível, levando a desafios no diagnóstico, além disso há diferentes métodos de realização de teste de suor, que dependem da experiência técnica de quem realiza, estando, portanto, sujeito a subjetividade de análise. Existe ainda a dificuldade particular de realizá-lo em bebês. Níveis intermediários de cloreto no suor (30-59 mmol/L) podem gerar confusão no diagnóstico, especialmente em bebês com uma ou nenhuma mutação causadora de FC.
A interpretação das mutações CFTR pode ser difícil, devido à falta de clareza sobre a relação entre genótipo e fenótipo, e à presença de genes modificadores e a definição de síndrome metabólica relacionada ao CFTR (CRMS) ou triagem de FC positiva. O diagnóstico inconclusivo (CFSPID) pode ser complexo e confuso. O acesso a serviços de referência específicos para pacientes com FC também pode ser difícil.
Conclusão
É importante educar profissionais de saúde e famílias sobre os desafios do diagnóstico precoce da FC e a importância do acompanhamento adequado.
Após o diagnóstico, os pais começam uma jornada em busca dos melhores cuidados e tratamentos, o que nem sempre é fácil. Pensando nisso, a Federação Europeia de Fibrose Cística (Cystic Fibrosis Europe) listou alguns passos para que os pais possam se preparar melhor para este processo.
- Informação:perguntar, estudar e ler sobre fibrose cística, usando fontes confiáveis. Certamente haverá muitas dúvidas, mas não se pode confiar em tudo o que está disponível na internet. Há muita informação desatualizada, já que os avanços em estudos e no tratamento da fibrose cística são contínuos. É recomendado que desenvolvam o hábito de anotar as dúvidas em um papel ou no computador e levem as anotações a cada visita ao médico.
- Apoio:buscar o envolvimento com associações e grupos de apoio aos pacientes e cuidadores. Conhecer e conversar com outras pessoas que estão passando ou já passaram pela mesma situação: ninguém melhor do que eles para ajudar. Os pais devem também falar sobre a fibrose cística para a família e amigos. Quanto mais eles entenderem a doença, mais estarão aptos para ajudar.
- Planejamento:o início da vida com fibrose cística pode ser desafiador, há várias coisas que parecem complexas no início, mas que em pouco tempo tornam-se rotineiras. Por isso, é importante que os pais estejam familiarizados com todos os passos do tratamento. No início, é recomendado que anotem como deverá ser o passo a passo das atividades relacionadas à gestão da fibrose cística em meio à rotina diária.
- Força:provavelmente os pais e familiares ouvirão palavras desanimadoras de pessoas bem intencionadas, mas mal informadas. Nestes momentos, devem lembrar que a FC costumava ser o que já não é mais, apesar de muitas pessoas ainda não saberem disso. Os pais devem estar preparados para encontrar negatividade, mas para manter este tipo de informação em perspectiva. Quando estiverem confusos, devem procurar ouvir os profissionais de saúde em quem confiam.